1. Preliminary analysis - SC#
The pre-processing workflows extract clean s-reads using tools below which can then be provided to the alignment applications and other downstream workflows similar to those used to analyze Isoseq data. High level pre-processing is adopted from Pacbio’s CLI workflow
skera for deconcatenating the MAS arrays into individual cDNA molecules and generate segmented reads (s-reads),
lima for removal of primers and identification of barcodes and orienting sequences in 5’ → 3’ orientation.
isoseq tag to clip tags (cell barcode, UMI, Gs). Supports design presets and custom experimental designs.
isoseq refine for trimming poly(A) tails and extracting Full length non-concatemer reads (FLNC) from s-reads.
isoseq correct for correcting errors in cell barcodes, the total number of usable reads increased (typically ~5%)
1.1. pbskera#
The pbskera workflow, as detailed below, processes raw HiFi reads generated with Sequel2e and Revio Long Read sequencers. The HiFi reads are a current default, and can be plugged in directly into the workflow to get segmented s-reads.
Workflow configuration for runnning these over cloud platforms supporting Cromwell like Terra can be found here:-
Dockstore : skera_w_QCplots.wdlGithub: Kinnex Preliminary ProcessingTest Data can be found here (public, requester-pays) : gs://fc-secure-6b69ce23-e507-4375-929c-75ab7213f277/kinnex_sc/m84014_240128_083549_s3_sub0005.hifi_reads.bcM0003.bam
The direct command executed here is:
1 skera split –j 16 reads.hifi.bam mas16_adapters.fasta reads.skera.bam
Input arguments for pbskera_main
Option name |
example value |
description |
|---|---|---|
input_bam |
this.input_bam |
Replace “input_bam” with the column that contains the path to the input file in the sample table. |
arraysize |
8 |
Size of the MAS array, could be 8, 16 or 12 depending upon the library type |
mas_adapters_fasta |
“gs://mdl-preprocess-refs/MAS_adapters/mas8_primers.fasta” |
MAS adapters FASTA file for de-concatenation. Ref files can be found at gs://mdl-preprocess-refs/MAS_adapters/ |
sample_id |
this.movie_name |
Replace “sample_id” with the column that contains the name of the samples in the sample table. Recommended id is the movie name |
gcs_output_dir |
“gs://output_dir/” |
Output directory to organize intermediates and QC plots |
num_threads |
8 |
Number of threads (set atleast equal to the number of cpu) |
cpu |
8 |
Number of cpus |
Example of QC plots generated :
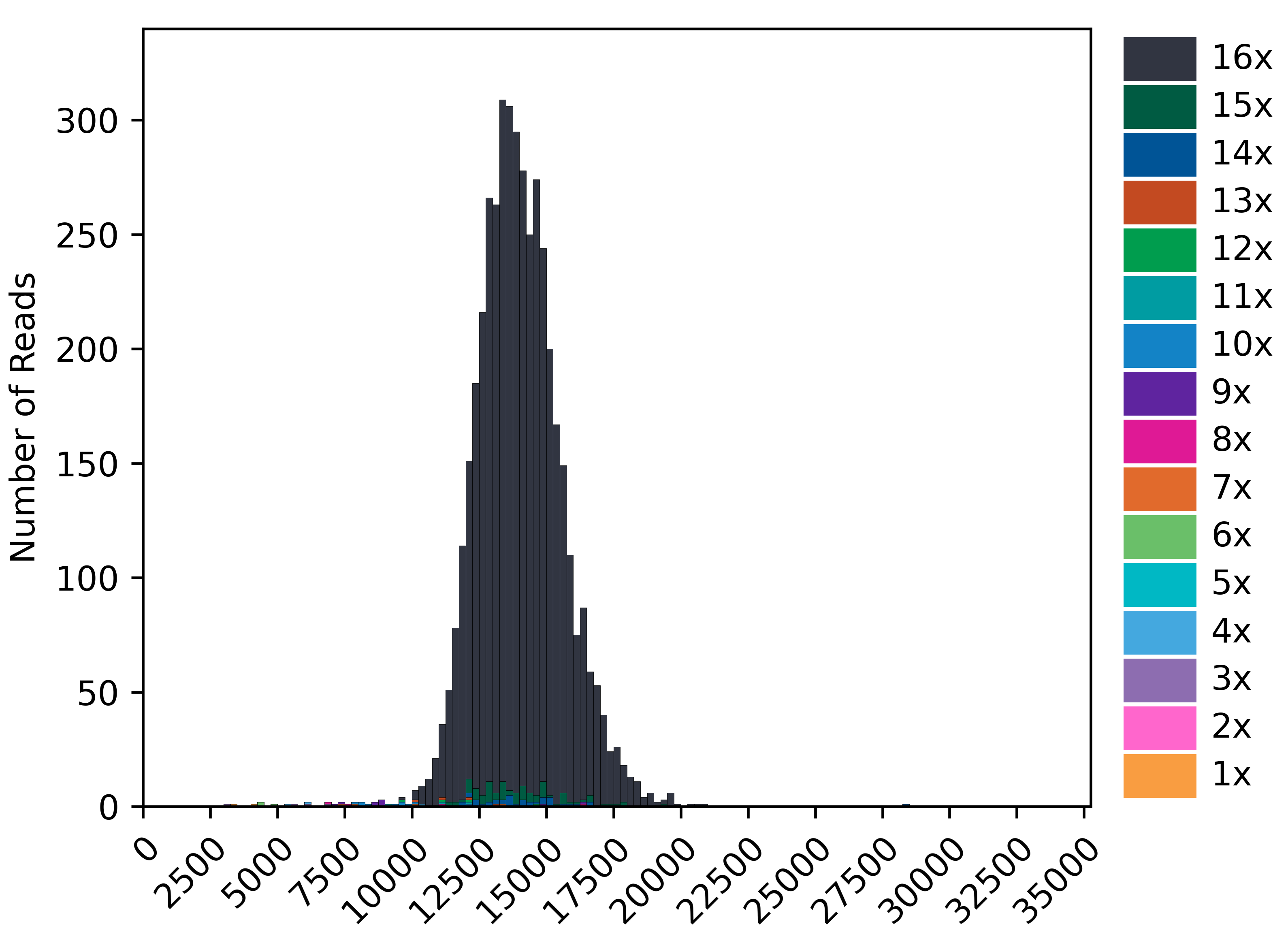

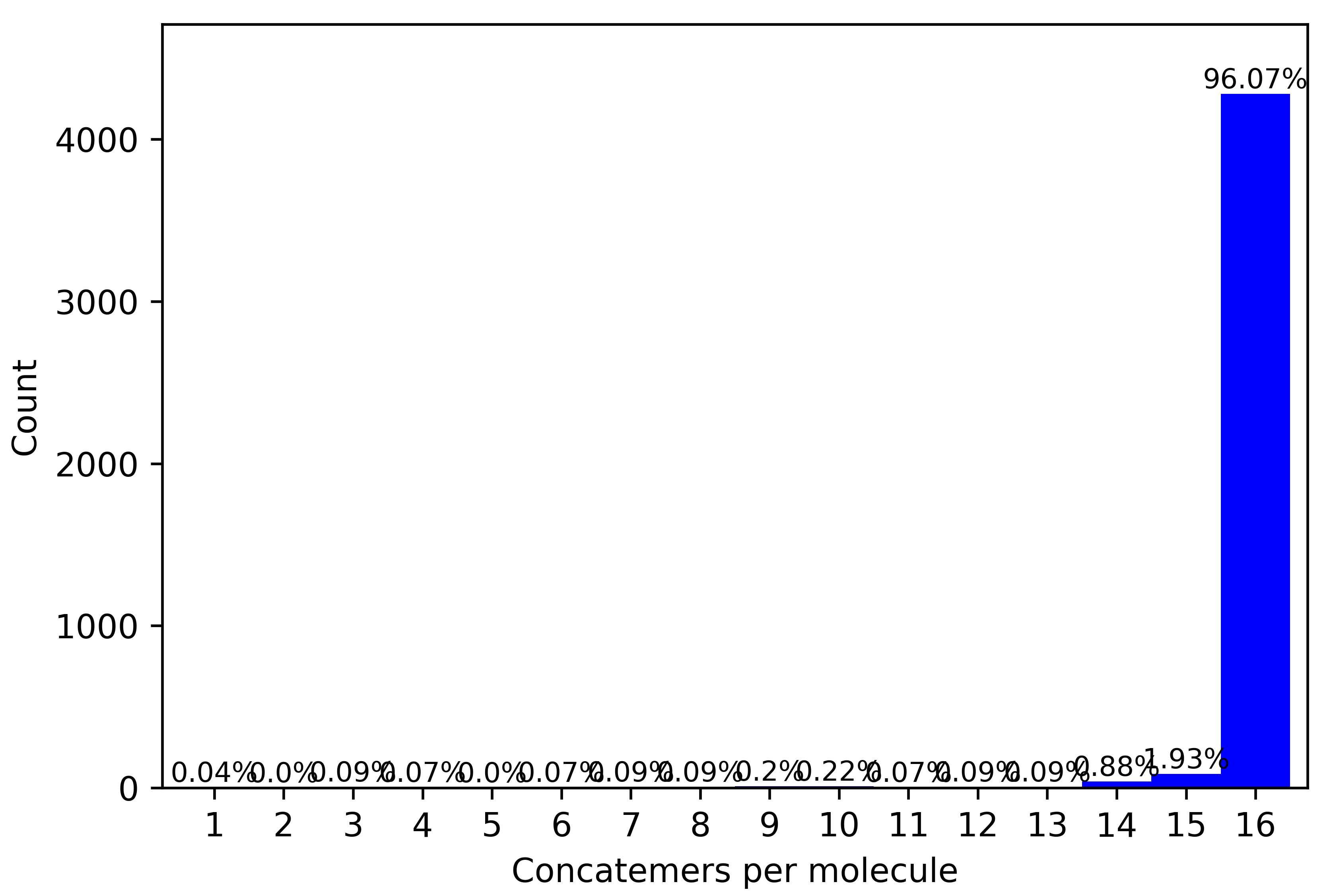
For a 16-mer we expect the plots to be similar to as above, with maximum number of reads assigned to a complete 16-mer configuration. In addition, to the readlength plot, the concatenation histogram should also indicate high percentages (>90%) to be assigned to a concatenation factor of 16. The ligation heatmap distributes the number of reads by adapter pairs found in the array. They should cleanly align along the diagonal for a well-performing array.
1.2 pblima + isoseq tag + isoseq refine + isoseq correct#

This workflow uses 2 tools to extract clean s-reads from the skera.bam received above namely lima and isoseq.
Workflow configuration for runnning these over cloud platforms supporting Cromwell like Terra can be found here:-
Dockstore : `sc_kinnex_lima_plus_isoseq.wdl <>`_Test Data: gs://fc-secure-6b69ce23-e507-4375-929c-75ab7213f277/kinnex_sc/m84014_240128_083549_s3_sub0005.bcM0003.skera.bam (public, requester-pays)
Example of input arguments for the workflow for 10x 3p kit
1{
2 "pb_sc_lima_isoseq.sample_id": "${this.movie_name}",
3 "pb_sc_lima_isoseq.barcodes_list": "gs://mdl-preprocess-refs/10x_barcodes/3M-february-2018-REVERSE-COMPLEMENTED.txt.gz",
4 "pb_sc_lima_isoseq.primer_fasta": "gs://mdl-preprocess-refs/REF-10x_primers/10x_3kit_primers.fasta",
5 "pb_sc_lima_isoseq.gcs_output_dir": "${this.out_path}",
6 "pb_sc_lima_isoseq.skera_bam": "${this.skera_bam}",
7 "pb_sc_lima_isoseq.read_design": "T-12U-16B"
8 }